Arq. Bras. Cardiol. 2025; 122(7): e20250825
Screening for Lysosomal Acid Lipase Deficiency in a Lipid Clinic
Zenia Brasil
![]() , Francisco Antonio H. Fonseca, Marco Antonio Curiati
, Francisco Antonio H. Fonseca, Marco Antonio Curiati
![]() , Sandra Obikawa Kyosen, Vanessa Gonçalves Pereira, Waleria Toledo Fonzar
, Sandra Obikawa Kyosen, Vanessa Gonçalves Pereira, Waleria Toledo Fonzar
![]() , João Bosco Pesquero, Francy Reis da Silva Patrício, Marcelo Hideki Yamamoto
, João Bosco Pesquero, Francy Reis da Silva Patrício, Marcelo Hideki Yamamoto
![]() , Joyce Umbelino Yamamoto, Vania D’Almeida, Ana Maria Martins, Maria Cristina Izar
, Joyce Umbelino Yamamoto, Vania D’Almeida, Ana Maria Martins, Maria Cristina Izar
![]()
This Original Article is referred by the Short Editorial "Beyond Cholesterol and Triglycerides in the Lipid Clinic: The Challenging Task of Identifying Lysosomal Acid Lipase Deficiency".
Abstract
Background
Lysosomal acid lipase deficiency (LAL-D) is a rare autosomal recessive disease, with massive accumulation of cholesteryl esters and triglycerides in many organs, leading to hepatosplenomegaly, microvesicular steatosis, cirrhosis and premature death. Early recognition is crucial for timely enzyme replacement therapy.
Objectives
To screen for LAL-D in subjects with dyslipidemias and/or liver disease at an outpatient lipid clinic.
Methods
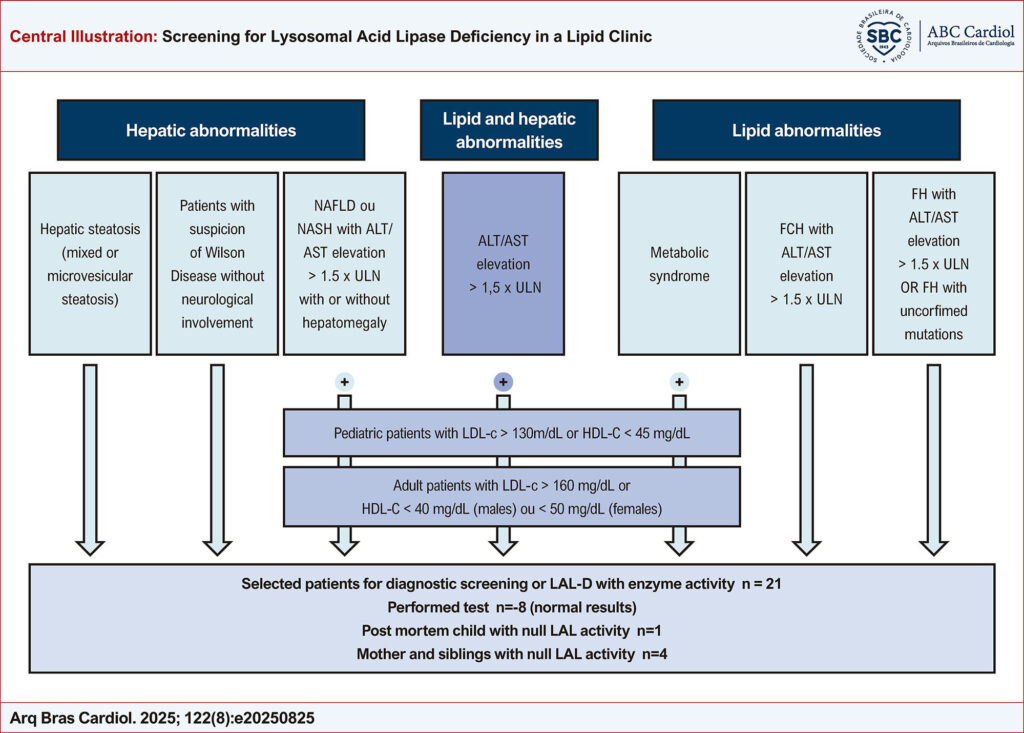
We retrospectively assessed records from 2,018 adults and children using a screening algorithm including ALT/AST elevation >1.5 x upper limit of normality, LDL-C>160 mg/dL, HDL-C<40 (males) or <50 mg/dL (females) in adults, and LDL-C>130 mg/dL, HDL-C<45 mg/dL, in children. High-risk patients for LAL-D were selected for LAL enzymatic activity assay in dried blood spots using LAL inhibitor, Lalistat-2.
Results
Among 2,018 screened patients, 21 (0.92%) were selected for LAL activity test, but only eight performed the test with normal results [mean LAL activity 0.077 ± 0.03 nmol/punch/h (reference value >0.024 nmol/punch/h)]. A child whose mother did not perform the test, had a post-mortem undetectable LAL activity. Further, the mother and three half-brothers confirmed LAL-D. Sequencing (NGS) of LIPA gene did not find pathogenic variants, not allowing to discard changes in non-coding region of the gene analyzed.
Conclusions
Identifying LAL-D remains a challenge, and an algorithm based on clinical and laboratory criteria may assist in selecting patients for LAL-D screening. Given its rarity and overlapping features with other genetic dyslipidemias, LAL-D is primarily a diagnosis of exclusion, often considered when other conditions have been ruled out.
Keywords: Dyslipidemias; Hepatomegaly; Lipase; Splenomegaly
180