Arq. Bras. Cardiol. 2025; 122(9): e20240847
Transcriptome High-Throughput Sequencing Analysis of lncRNA and mRNA Expression in Patients with Coronary Slow Flow
Haibing Jiang
![]() , Yi Yang, Xueqin Zhai, Lijing Zhang, Aerziya Kahaerjiang
, Yi Yang, Xueqin Zhai, Lijing Zhang, Aerziya Kahaerjiang
This Original Article is referred by the Short Editorial "Functional RNA Applications in Cardiovascular Precision Medicine: Advances and Diagnostic Perspectives".
Abstract
Background
Long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) are believed to play key roles in the pathophysiology of coronary slow flow (CSF).
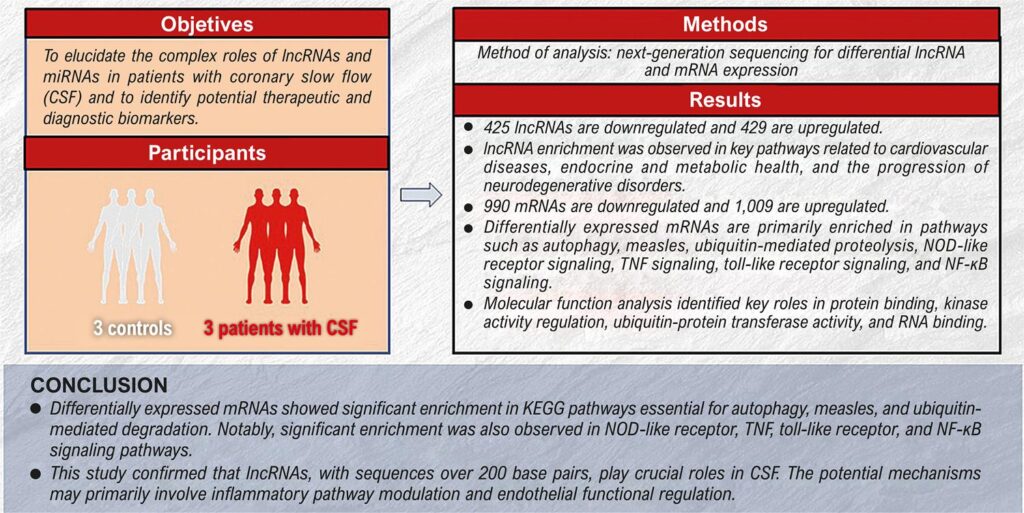
Objectives
This study aimed to explore the complex biological networks involved in CSF using whole-transcriptome sequencing, with the goal of identifying potential diagnostic biomarkers and therapeutic targets.
Methods
Whole-transcriptome sequencing was performed on samples from three patients with CSF and three matched control subjects. A p-value < 0.05 was considered statistically significant.
Results
A total of 854 lncRNAs were differentially expressed, with 425 downregulated and 429 upregulated. KEGG pathway analysis showed significant enrichment of lncRNAs in pathways associated with cardiovascular diseases, endocrine and metabolic disorders, and neurodegenerative disease progression. Additionally, 1,999 mRNAs were differentially expressed, including 990 downregulated and 1,009 upregulated. Molecular function analysis identified roles in protein binding, regulation of kinase activity, ubiquitin-protein transferase activity, and RNA binding. KEGG analysis indicated that the differentially expressed mRNAs were primarily involved in autophagy, measles, ubiquitin-mediated proteolysis, the NOD-like receptor signaling pathway, the tumor necrosis factor (TNF) signaling pathway, the toll-like receptor (TLR) signaling pathway, and the NF-κB signaling pathway.
Conclusions
Differentially expressed mRNAs were significantly enriched in KEGG pathways related to autophagy, measles, and ubiquitin-mediated degradation, as well as in signaling cascades involving NOD-like receptors, TNF, TLR, and NF-κB. Further studies are required to validate these findings.
158